
Effects of ring-strain on the ultrafast photochemistry of cyclic ketones†

Abstract
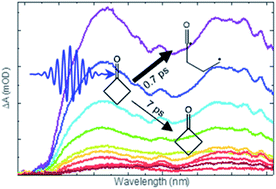
Ring-strain in cyclic organic molecules is well-known to influence their chemical reactivity. Here, we examine the consequence of ring-strain for competing photochemical pathways that occur on picosecond timescales. The significance of Norrish Type-I photochemistry is explored for three cyclic ketones in cyclohexane solutions at ultraviolet (UV) excitation wavelengths from 255–312 nm, corresponding to an π* ← n excitation to the lowest excited singlet state (S1). Ultrafast transient absorption spectroscopy with broadband UV/visible probe laser pulses reveals processes common to cyclobutanone, cyclopentanone and cyclohexanone, occurring on timescales of ≤1 ps, 7–9 ps and >500 ps. These kinetic components are respectively assigned to prompt cleavage of an α C–C bond in the internally excited S1-state molecules prepared by UV absorption, vibrational cooling of these hot-S1 molecules to energies below the barrier to C–C bond cleavage on the S1 state potential energy surface (with commensurate reductions in the energy-dependent α-cleavage rate), and slower loss of thermalized S1-state population. The thermalized S1-state molecules may competitively decay by activated reaction over the barrier to α C–C bond fission on the S1-state potential energy surface, internal conversion to the ground (S0) electronic state, or intersystem crossing to the lowest lying triplet state (T1) and subsequent C–C bond breaking. The α C–C bond fission barrier height in the S1 state is significantly reduced by the ring-strain in cyclobutanone, affecting the relative contributions of the three decay time components which depend systematically on the excitation energy above the S1-state energy barrier. Transient infra-red absorption spectra obtained after UV excitation identify ring-opened ketene photoproducts of cyclobutanone and their timescales for formation.
 Please wait while we load your content...
Please wait while we load your content...

