
Understanding the structures and aromaticity of heteroporphyrins with computations†
 *a
and
Yuan-Bin
She
*a
*a
and
Yuan-Bin
She
*a
Abstract
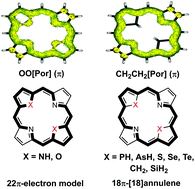
Heteroporphyrins are porphyrin derivatives with replacement of the pyrrolic NH moiety by other heteroatom-containing groups, such as PH, AsH, SiH2, O, S, etc. For all studied heteroporphyrins, the macrocycle structure is distorted due to the presence of large heteroatoms. The HOMO–LUMO gap of heteroporphyrins is generally decreased compared to regular porphyrins. Both nucleus independent chemical shifts values and visualized anisotropy of induced current density were computed to describe the aromaticity of heteroporphyrins. The plots of anisotropy of induced current density suggest that the ring current diverged into an outer and an inner pathway at each ring. The current mainly passes through the outer path at the pyrrolic rings with inner hydrogen and through the inner path at the pyrrolic rings without inner hydrogen. In both regular porphyrin and O-substituted heteroporphyrins, the aromatic pathway is mainly contributed by the 22π-electron aromatic route model. Heteroatoms such as PH, AsH, S, Se and Te have little contribution to the aromaticity of heteroporphyrins. In addition, the π conjugation is also interrupted at the CH2 and SiH2 moiety, and the ring current mainly passes through the outer path of the heteroporphyrins with CH2 and SiH2 replacing the pyrrolic NH moiety. Therefore the 18π-[18]annulene model is dominated in PH-, AsH-, S-, Se-, Te-, CH2- and SiH2-substituted heteroporphyrins. These computational studies shed new light on the aromatic characters of heteroporphyrins, and will facilitate the further development of various novel heteroporphyrins.
- This article is part of the themed collection: Mechanistic, computational & physical organic chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

