
Synthesis, radiolabelling and initial biological characterisation of 18F-labelled xanthine derivatives for PET imaging of Eph receptors†‡
 §¶ab
Christin
Neuber,
§a
Martin
Köckerling,
c
Amedeo
Caflisch,
d
Jörg
Steinbach,
ab
Jens
Pietzsch
ab
and
Constantin
Mamat
*ab
§¶ab
Christin
Neuber,
§a
Martin
Köckerling,
c
Amedeo
Caflisch,
d
Jörg
Steinbach,
ab
Jens
Pietzsch
ab
and
Constantin
Mamat
*ab
Abstract
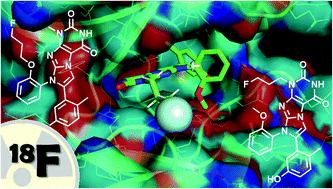
Eph receptor tyrosine kinases, particularly EphA2 and EphB4, represent promising candidates for molecular imaging due to their essential role in cancer progression and therapy resistance. Xanthine derivatives were identified to be potent Eph receptor inhibitors with IC50 values in the low nanomolar range (1–40 nm). These compounds occupy the hydrophobic pocket of the ATP-binding site in the kinase domain. Based on lead compound 1, we designed two fluorine-18-labelled receptor tyrosine kinase inhibitors ([18F]2/3) as potential tracers for positron emission tomography (PET). Docking into the ATP-binding site allowed us to find the best position for radiolabelling. The replacement of the methyl group at the uracil residue ([18F]3) rather than the methyl group of the phenoxy moiety ([18F]2) by a fluoropropyl group was predicted to preserve the affinity of the lead compound 1. Herein, we point out a synthesis route to [18F]2 and [18F]3 and the respective tosylate precursors as well as a labelling procedure to insert fluorine-18. After radiolabelling, both radiotracers were obtained in approximately 5% radiochemical yield with high radiochemical purity (>98%) and a molar activity of >10 GBq μmol−1. In line with the docking studies, first cell experiments revealed specific, time-dependent binding and uptake of [18F]3 to EphA2 and EphB4-overexpressing A375 human melanoma cells, whereas [18F]2 did not accumulate at these cells. Since both tracers [18F]3 and [18F]2 are stable in rat blood, the novel radiotracers might be suitable for in vivo molecular imaging of Eph receptors with PET.
- This article is part of the themed collection: Chemical Biology in OBC
 Please wait while we load your content...
Please wait while we load your content...

