
Mechanism and stereoselectivity of benzylic C–H hydroxylation by Ru–porphyrin: a computational study†

Abstract
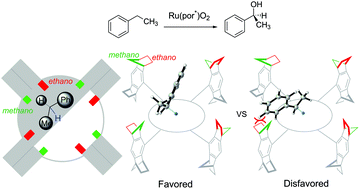
The mechanism and origin of the stereoselectivity of asymmetric benzylic C–H hydroxylation by Ru–porphyrin were elucidated with density functional theory calculations. The reaction proceeds via a hydrogen-atom abstraction/oxygen-rebound pathway, wherein a high-valent ruthenium-oxo species abstracts a hydrogen atom from ethylbenzene to generate a radical pair intermediate, followed by the oxygen-rebound process to form 1-phenylethanol. The hydrogen-atom abstraction step is the rate- and stereoselectivity-determining step. Based on the mechanistic model, the computed stereoselectivity is in agreement with the experimental observations. Analysis of the distortion/interaction model suggests that stereoselectivity is determined by both the distortion energy of the ethylbenzene and the interaction energy between the ethylbenzene and the chiral Ru–porphyrin. The steric repulsion between the phenyl group of ethylbenzene and the bulky substituent of Ru–porphyrin is the leading cause of chiral induction.
- This article is part of the themed collection: Mechanistic, computational & physical organic chemistry in OBC
 Please wait while we load your content...
Please wait while we load your content...

