
Photocatalytic hydrogen evolution using a Ru(ii)-bound heteroaromatic ligand as a reactive site†
 a
Dachao
Hong,
ab
Hiroaki
Kotani,
a
Kazunari
Yoshizawa,
*c
Shunichi
Fukuzumi
de
and
Takahiko
Kojima
*a
a
Dachao
Hong,
ab
Hiroaki
Kotani,
a
Kazunari
Yoshizawa,
*c
Shunichi
Fukuzumi
de
and
Takahiko
Kojima
*a
Abstract
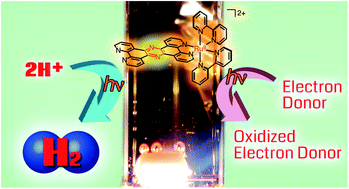
A RuII complex, [RuII(tpphz)(bpy)2]2+ (1) (tpphz = tetrapyridophenazine, bpy = 2,2′-bipyridine), whose tpphz ligand has a pyrazine moiety, is converted efficiently to [RuII(tpphz-HH)(bpy)2]2+ (2) having a dihydropyrazine moiety upon photoirradiation of a water–methanol mixed solvent solution of 1 in the presence of an electron donor. In this reaction, the triplet metal-to-ligand charge-transfer excited state (3MLCT*) of 1 is firstly formed upon photoirradiation and the 3MLCT* state is reductively quenched with an electron donor to afford [RuII(tpphz˙−)(bpy)2]+, which is converted to 2 without the observation of detectable reduced intermediates by nano-second laser flash photolysis. The inverse kinetic isotope effect (KIE) was observed to be 0.63 in the N–H bond formation of 2 at the dihydropyrazine moiety. White-light (380–670 nm) irradiation of a solution of 1 in a protic solvent, in the presence of an electron donor under an inert atmosphere, led to photocatalytic H2 evolution and the hydrogenation of organic substrates. In the reactions, complex 2 is required to be excited to form its 3MLCT* state to react with a proton and aldehydes. In photocatalytic H2 evolution, the H–H bond formation between photoexcited 2 and a proton is involved in the rate-determining step with normal KIE being 5.2 on H2 evolving rates. Density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations on the reaction mechanism of H2 evolution from the ground and photo-excited states of 2 were performed to have a better understanding of the photocatalytic processes.
- This article is part of the themed collection: Spotlight Collection: Photoinduced redox chemistry
 Please wait while we load your content...
Please wait while we load your content...

