
Conformational dynamics of aqueous hydrogen peroxide from first principles molecular dynamics simulations
 a
and
Bhabani S.
Mallik
*a
a
and
Bhabani S.
Mallik
*a
Abstract
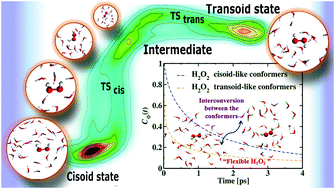
We performed first principles molecular dynamics simulations of a relatively dilute aqueous hydrogen peroxide (H2O2) solution to examine its structural alterations and relevant dynamics upon solvation. The internal rotation of the OH groups about the O–O bond facilitates the flexible structure of H2O2. Structural calculations reveal dihedral angle fluctuations in the aqueous solution. Water molecules make stronger hydrogen bonds through the hydrogen atom of the solute than the oxygen atom leading to distinct hydrogen bonding configurations inside the first solvation shell. Time-dependent dihedral angle alterations result in conformational changes and the normalized dihedral angle distribution plot displays characteristic peaks at ∼100–120° and ∼230°, illustrating various conformational states. Within the simulation time, flexibility-induced interconversion of hydrogen peroxide gives rise to several cisoid and transoid conformers. In this study, we examine the relative population of the associated conformational states and the lifetime of the cisoid and transoid conformers from the torsion angle variations. We also determine the free energy landscape of the rotational isomerization process in H2O2 and explore two distinct energy barriers during such interconversion.
 Please wait while we load your content...
Please wait while we load your content...

