
Using coarse-grained molecular dynamics to rationalize biomolecule solubilization mechanisms in ionic liquid-based colloidal systems†

Abstract
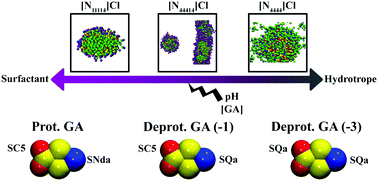
Solubilizing agents are widely used to extract poorly soluble compounds from biological matrices. Aqueous solutions of surfactants and hydrotropes are commonly used as solubilizers, however, the underlying mechanism that determines their action is still roughly understood. Among these, ionic liquids (IL) are often used not only for solubilization of a target compound but in liquid–liquid extraction processes. Molecular dynamics simulations can shed light into this issue by providing a microscopic insight of the interactions between solute and solubilising agents. In this work, a new coarse-grained (CG) model was developed under the MARTINI framework for gallic acid (GA) while the CG models of three quaternary ammonium ionic liquids and salts (QAILS) were obtained from literature. Three QAILS were selected bearing in mind their potential solubilising mechanisms: trimethyl-tetradecylammonium chloride ([N1,1,1,14]Cl) as a surfactant, tetrabutylammonium chloride ([N4,4,4,4]Cl) as a hydrotrope, and tributyl-tetradecylammonium chloride ([N4,4,4,14]Cl) as a system combining the characteristics of the other compounds. Throughout this hydrotrope-to-surfactant spectrum and considering the most prevalent GA species across the pH range, the solvation of GA at two concentration levels in aqueous QAILS solutions were studied and discussed. The results of this study indicate that dispersive interactions between the QAILS and GA are generally the driving force in the GA solubilization. However, electrostatic interactions play an increasingly significant role as the GA becomes deprotonated, affecting their placement within the micelle and ultimately the solvation mechanism. The hydrotropic mechanism seen in [N4,4,4,4]Cl corroborates recent models based on the formation of a hydrotrope-solute aggregates driven by dispersive forces. This work contributes to the application of a transferable approach to partition and solubilization studies using molecular dynamics, which could complement experimental assays and quickly screen molecular candidates for these processes.
 Please wait while we load your content...
Please wait while we load your content...

