
Energetics of non-heme iron reactivity: can ab initio calculations provide the right answer?†

Abstract
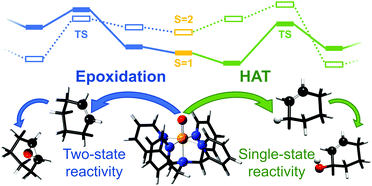
We use a variety of computational methods to characterize and compare the hydrogen atom transfer (HAT) and epoxidation reaction pathways for oxidation of cyclohexene by an iron(IV)-oxo complex. Previous B3LYP calculations have led to predictions that both alcohol (from the HAT route) and epoxide should be formed in similar amounts, which was not in agreement with experiment where only the HAT product was observed. We show here that ab initio calculations which can take both static and dynamic correlation into account are needed to explain the experimentally observed dominance of the HAT process. Since these systems do not have very strong multireference character we have also tested different flavours of local coupled cluster methods. We suggest that further improvements are necessary before they can provide highly accurate results for these systems.
 Please wait while we load your content...
Please wait while we load your content...

