
Quantifying how step-wise fluorination tunes local solute hydrophobicity, hydration shell thermodynamics and the quantum mechanical contributions of solute–water interactions†
 ‡a
Denilson
Mendes de Oliveira,
‡b
Petra
Imhof,
§c
Dor
Ben-Amotz
b
and
Ana
Vila Verde
¶*a
‡a
Denilson
Mendes de Oliveira,
‡b
Petra
Imhof,
§c
Dor
Ben-Amotz
b
and
Ana
Vila Verde
¶*a
Abstract
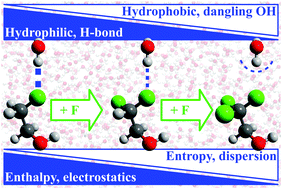
The ability to locally tune solute–water interactions and thus control the hydrophilic/hydrophobic character of a solute is key to control molecular self-assembly and to develop new drugs and biocatalysts; it has been a holy grail in synthetic chemistry and biology. To date, the connection between (i) the hydrophobicity of a functional group; (ii) the local structure and thermodynamics of its hydration shell; and (iii) the relative influence of van der Waals (dispersion) and electrostatic interactions on hydration remains unclear. We investigate this connection using spectroscopic, classical simulation and ab initio methods by following the transition from hydrophile to hydrophobe induced by the step-wise fluorination of methyl groups. Along the transition, we find that water–solute hydrogen bonds are progressively transformed into dangling hydroxy groups. Each structure has a distinct thermodynamic, spectroscopic and quantum-mechanical signature connected to the associated local solute hydrophobicity and correlating with the relative contribution of electrostatics and dispersion to the solute–water interactions.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

