
Attenuated total reflectance far-ultraviolet and deep-ultraviolet spectroscopy analysis of the electronic structure of a dicyanamide-based ionic liquid with Li+†

Abstract
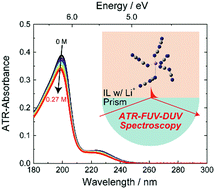
The electronic states of N-butyl-N-methylpyrrolidinium dicyanamide ([BMP][DCA]), a solvated ionic liquid, around Li+ were investigated using attenuated total reflectance far-ultraviolet and deep-ultraviolet (ATR-FUV-DUV) spectroscopy. The absorption bands ascribed to the [DCA]− were blue-shifted as the Li+ concentration increased, and the origin of the shift was explained by the energetic destabilization of the final (excited) molecular orbital using time-dependent density functional theory (TD-DFT) calculations. Using the multivariate curve resolution-alternating least squares (MCR-ALS) algorithm, the obtained spectra were decomposed into two types of [DCA]− at electronic state level, which were categorised as pure [BMP][DCA] and [DCA]− affected by Li+. Our results revealed that the number of [DCA]− with electronic states affected by a Li+, which was termed the electronic coordination number, was ∼5. This value was different from the coordination number within the first solvation layer, which was ∼4. Combining the TD-DFT with molecular dynamics simulations, we demonstrated that one [DCA]− outside the first solvation layer had a different electronic state from that of pure [BMP][DCA]. This is the first successful study that combines ATR-FUV-DUV spectroscopy with MCR-ALS calculations to build a solvation model that describes the electronic states.
 Please wait while we load your content...
Please wait while we load your content...

