
Solvation energies of ions with ensemble cluster-continuum approach†
 a
Petr
Slavíček
*a
a
Petr
Slavíček
*a
Abstract
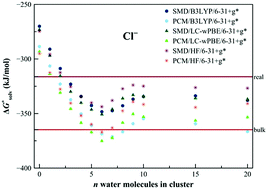
Solvation free energies can be advantageously estimated by cluster-continuum approaches. They proved useful especially for systems with high charge density. However, the clusters are assumed to be single minimum rigid species. It is an invalid condition for larger clusters and it complicates the assessment of convergence with the system size. We present a new variant of the cluster-continuum approach, “Ensemble Cluster-Continuum” scheme, where the single minima problem is circumvented by a thermodynamic cycle based on vertical quantities (ionization energies, electron affinities). Solvation free energies are calculated for a charged-neutralized system and solvation correction for the vertical quantities is estimated for an ensemble of structures from molecular dynamics simulation. We test the scheme on a set of various types of anions and cations, we study the convergence of the cluster-continuum model and assess various types of errors. The quantitative data depend on the applied continuum solvation model yet the convergence is analogous. We argue that the assessment of convergence provides a measure of the reliability of the calculated solvation energies.
 Please wait while we load your content...
Please wait while we load your content...

