
Atomistic simulations of the aggregation of small aromatic molecules in homogenous and heterogenous mixtures†
 b
Li-Juan
Yu,
cd
Amir
Karton,
c
Graham S.
Chandler,
c
Dahbia
Talbi
*f
and
Dino
Spagnoli
*c
b
Li-Juan
Yu,
cd
Amir
Karton,
c
Graham S.
Chandler,
c
Dahbia
Talbi
*f
and
Dino
Spagnoli
*c
Abstract
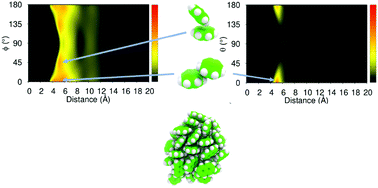
The relatively weak London dispersion forces are the only interactions that could cause aggregation between simple aromatic molecules. The use of molecular dynamics and high-level ab initio computer simulations has been used to describe the aggregation and interactions between molecular systems containing benzene, naphthalene and anthracene. Mixtures containing one type of molecule (homogenous) and more than one type of molecule (heterogenous) were considered. Our results indicate that as molecular weight increases so does the temperature at which aggregation will occur. In all simulations, the mechanism of aggregation is through small clusters coalescing into larger clusters. The structural analysis of the molecules within the clusters reveals that benzene will orient itself in T-shaped and parallel displaced configurations. Molecules of anthracene prefer to orient themselves in a similar manner to a bulk crystal with no T-shaped configuration observed. The aggregation of these aromatic molecules is discussed in the context of astrochemistry with particular reference to the dust formation region around stars.
 Please wait while we load your content...
Please wait while we load your content...

