
Computational mechanistic investigation of the Fe + CO2 → FeO + CO reaction†

Abstract
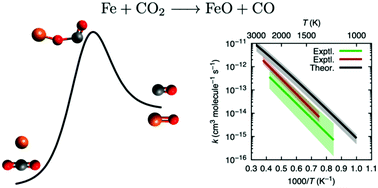
We report a computational study of the mechanism and determination of the rate constants of the Fe + CO2 → FeO + CO reaction, in the 1000–3000 K temperature range, at the CCSD(T)/CBS//B3LYP/def2-TZVP level of theory. The overall rate constant was obtained by a Kinetic Monte Carlo simulation. The calculated rate constant, at 2000 K, is 9.72 × 10−13 cm3 molecule−1 s−1, in agreement with experimental measurements: 2.97 × 10−13 cm3 molecule−1 s−1 [A. Giesen et al., Phys. Chem. Chem. Phys., 2002, 4, 3665] and 1.13 × 10−13 cm3 molecule−1 s−1 [V. N. Smirnov, Kinet. Catal., 2008, 49, 607]. Our study shows that this reaction follows a complex mechanism, with multiple reaction paths contributing to the overall rate, and that CCSD(T) accurately describes this transition metal reaction.
 Please wait while we load your content...
Please wait while we load your content...

