
Including dispersion in density functional theory for adsorption on flat oxide surfaces, in metal–organic frameworks and in acidic zeolites†

Abstract
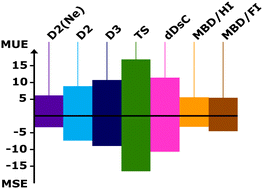
We examine the performance of nine commonly used methods for including dispersion interactions in density functional theory (DFT): three different parametrizations of damped 1/Rn terms (n = 6, 8, …) added to the DFT energy (Grimme's D2 and D3 parameterizations as well as that of Tkatchenko and Scheffler), three different implementations of the many-body dispersion approach (MBD, MBD/HI and MBD/FI), the density-dependent energy correction, called dDsC, and two “first generation” van der Waals density functionals, revPBE-vdW and optB86b-vdW. As test set we use eight molecule–surface systems for which agreement has been reached between experiment and hybrid QM:QM calculations within chemical accuracy limits (±4.2 kJ mol−1). It includes adsorption of carbon monoxide and dioxide in the Mg2(2,5-dioxido-1,4-benzenedicarboxylate) metal–organic framework (Mg-MOF-74, CPO-27-Mg), adsorption of carbon monoxide as well as of monolayers of methane and ethane on the MgO(001) surface, as well as adsorption of methane, ethane and propane in H-chabazite (H-CHA). D2 with Ne parameters for Mg2+, D2(Ne), MBD/HI and MBD/FI perform best. With the PBE functional, the mean unsigned errors are 6.1, 5.6 and 5.4 kJ mol−1, respectively.
- This article is part of the themed collection: Bunsentagung 2020: Understanding Dispersion Interactions in Molecular Chemistry
 Please wait while we load your content...
Please wait while we load your content...

