
Toward an understanding of electronic excitation energies beyond the molecular orbital picture†
 a
and
Felix
Plasser
*a
a
and
Felix
Plasser
*a
Abstract
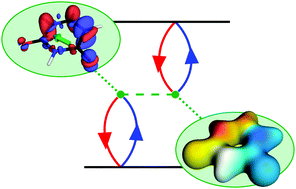
Tuning the energies of molecular excited states is a central research theme in modern chemistry with high relevance for optoelectronic applications and chemical synthesis. Whereas frontier orbitals have proven to be an intuitive and simple model in many cases, they can only provide a very rough approximation of the underlying wavefunctions. The purpose of this Perspective is to explore how our qualitative understanding of electronic excitation processes can be promoted beyond the molecular orbital picture by exploiting methods and insights from modern quantum chemistry. For this purpose, the physics of a correlated electron–hole pair is analysed in detail to show the origin of exchange repulsion and a dynamic Coulomb attraction, which determine its energy aside from the orbital energies. Furthermore, we identify and discuss the two additional effects of secondary orbital relaxation and de-excitations. Rules for reconstructing these four contributions from general excited-state computations are presented and their use is exemplified in three case studies concerned with the relative ordering of the singlet and triplet ππ* and nπ* states of uracil, the large energetic differences between the first singlet and triplet states of the polyacenes, and the assignment of plasmonic states in octatetraene. Finally, we lay out some general ideas for how the knowledge gained could ultimately lead to new design principles for tuning molecular excitation energies as well as for diagnosing possible shortcomings of commonly used electronic structure methods.
- This article is part of the themed collection: PCCP Perspectives
 Please wait while we load your content...
Please wait while we load your content...

