
Quantum mechanical molecular dynamics simulations of polaron formation in methylammonium lead iodide perovskite†

Abstract
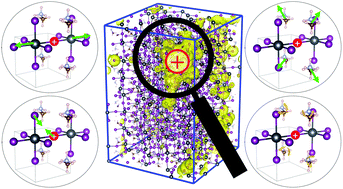
We investigated the atomistic and dynamical mechanism of polaron formation in methylammonium lead iodide perovskite (MAPbI3), which is a representative perovskite solar cell absorber, through the quantum mechanical molecular dynamics simulations. The simulations were conducted on the spatial scale of several nanometres, which can describe charge localization and the associated structural deformation, using the divide-and-conquer-type density-functional tight-binding method, which enables a quantum chemical treatment of systems comprising thousands of atoms. We found that both the structural parts of MAPbI3, namely, the inorganic framework (PbI3−) and the MA cations, involve the structural deformation associated with polaron formation. We elucidated that in the process of polaron formation, charge localization is invoked by thermal structural fluctuation, and a further structural deformation is caused by the relaxation of the charge carrier. Finally, importance of the two structural parts, PbI3− and MA, was examined from the energetical viewpoint.
- This article is part of the themed collection: 2019 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

