
Theoretical perspective for luminescent mechanism of thermally activated delayed fluorescence emitter with excited-state intramolecular proton transfer†
 a
Lili
Lin
*a
a
Lili
Lin
*a
Abstract
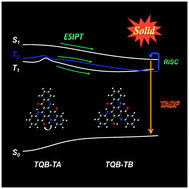
Excited-state intramolecular proton transfer (ESIPT) and thermally activated delayed fluorescence (TADF) are interesting photochemical and photophysical properties for light-emitting materials. Recently, a novel design strategy for TADF molecules was realized in the TQB molecule by ESIPT [ACS Cent. Sci., 2017, 3(7), 769–777]; however, the TADF and ESIPT mechanisms for this molecule are not very clear. In this work, the TADF and ESIPT mechanisms for TQB in both solution and the solid phase were studied based on the calculation of potential energy curves, energy levels and decay rates of the excited states. It was found that proton transfer in S1 for TQB is quite easy in both DMF and the solid phase. In contrast, the proton transfer in T1 and T2 in DMF is slightly difficult, while the barrier in T1 for TQB in the solid phase is comparable with room temperature energy. The T2TQB-TA → S1TQB-TB upconversion path plays a crucial role in the TADF of TQB in solvent, while T2TQB-TB → S1TQB-TB has a dominant contribution to the TADF in the solid phase. Based on the investigation of TQB, we designed two molecules, TQB* with a single hydrogen bond and TQB** with two hydrogen bonds. Our calculation results indicate that the TQB** molecules can realize dual emission by ESIPT and are a potential light-emitting material for white light. No ESIPT can be realized in TQB*, while large energy gaps are found between the higher excited states and lowest excited states. Our investigation provides new perspectives for the ESIPT and TADF mechanisms, which can help in understanding the light-emitting properties of TQB and provide some insights on the design of new functional luminescent molecules.
 Please wait while we load your content...
Please wait while we load your content...

