
Polarons in π-conjugated ladder-type polymers: a broken symmetry density functional description†

Abstract
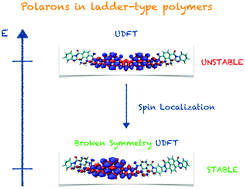
Electronic charged states (i.e., polarons) play a crucial role in governing charge transfer, spin, thermo-electric and redox mechanisms in organic functional materials. An accurate description at the quantum-chemical level is mandatory to understand their response and transport properties. We report a comprehensive computational investigation concerning the polaron properties of a high electron conductivity (n-type) π-conjugated ladder-type polymer, namely polybenzimidazobenzophenanthroline (BBL). We show how spin polarized unrestricted Density Functional Theory (UDFT) and restricted (RDFT) methods can lead to solutions of the polaron and bipolaron electronic wavefunctions which are not the most stable ones. This aspect can be traced back to the multiconfigurational character of the electronic charged states’ wavefunction. We demonstrate how broken symmetry DFT (BS-UDFT) can circumvent this issue, well describing the polaron/bipolaron localization in terms of spin densities and structural deformations, thus providing a correct assessment of the electron transport parameters (e.g., reorganization energy), otherwise incorrectly computed at the UDFT/RDFT levels. Our calculations are further validated by comparing the IR spectra of polaronic species with the experimental one, as measured on doped BBL films. Our study calls for an urgent and careful computational assessment of the electronic charged states (e.g., polaron, bipolaron, etc.), in high performance π-conjugated materials, such as ladder-type polymers and other donor–acceptor derivatives, for a correct understanding of their charge, heat, and spin transport mechanisms.
- This article is part of the themed collection: Journal of Materials Chemistry C Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

