
The crystal structure of visible light absorbing piezoelectric semiconductor SrNb2V2O11 revisited: high-resolution X-ray diffraction, vibrational spectroscopy and computational study†

Abstract
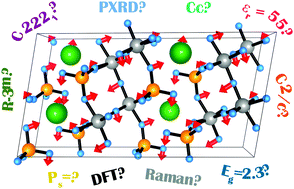
Ferroelectric materials have a long-term track record of applications in electronics due to their spontaneous electric polarization. This property can be coupled with photoabsorption properties, resulting in a bulk photoelectric effect, the new on-the-edge domain for ferroelectric use. In this sense, considering the low bandgap of binary strontium-niobium ortho-vanadate SrNb2V2O11, which has recently been reported as ferroelectric, we propose here a deep experimental and computational understanding of its structural and physical properties, considered relevant for further applications. Microcrystalline SrNb2V2O11 was prepared by a conventional solid state route, proposing a synthetic pathway deduced from thermoanalytical observations and high-temperature powder X-ray diffraction. The crystal structure (space group Cc, a = 18.15415(2) Å, b = 5.52811(6) Å, c = 9.52728(1) Å, β = 99.8033(8)°, Z = 2), successfully solved using high resolution powder X-ray diffraction, reveals the presence of distorted perovskite-like [Nb4V2O12] units when preparing [Nb2V2O11] sheets. By application of symmetry adapted mode analysis, the non-centrosymmetry originates from Sr atom displacements and [Nb4V2O12] unit “breathing” deformations, which can be explained in terms of the group–subgroup relationship. By ground state analysis of the polytypes across possible C-centered monoclinic cells, only the present experimentally based structural model (space group Cc) can be adopted, substituting the so far reported crystallographic data. The semiconducting nature of the phase, with a direct bandgap of 2.3 eV, was determined by optical absorption measurements and confirmed computationally. By coupling Raman spectroscopy and density functional perturbation theory, the dielectric properties (εriso = 55) were accurately calculated and the observed optical phonons were fully interpreted. Finally, using the Berry phase formalism, we predicted a value of spontaneous polarization of 16.6 μC cm−2 in the absence of confident existing experimental data.
 Please wait while we load your content...
Please wait while we load your content...

