
The distinctive phase stability and defect physics in CsPbI2Br perovskite†

Abstract
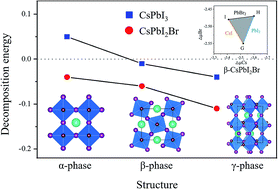
In this work, the structural, optoelectronic, and defect properties of both the α-phase and β-phase CsPbI2Br are investigated based on first-principles calculations. First, we calculate the bandgap of α-CsPbI2Br using the standard HSE06 method which is in accordance with the experimental bandgap value of 1.9 eV. We also find that α-CsPbI2Br shows comparable optoelectronic properties to CsPbI3. More intriguingly, our calculated results show that both α-phase and β-phase CsPbI2Br possess better thermal and phase stability than CsPbI3 due to the mixture of halide elements, and the optical absorption coefficient of CsPbI2Br also resembles that of CsPbI3 and MAPbI3 due to the lone pair electrons of Pb. Furthermore, the formation energies and transition energy levels of intrinsic point defects indicate that CsPbI2Br exhibits unipolar self-doping behavior (p-type conductivity) and similarly good defect tolerance while the dominant defect changes. Therefore, our present work provides fundamental understanding of the great performance of CsPbI2Br thin films and offers strategies for designing more superior CsPbI2Br-based devices.
 Please wait while we load your content...
Please wait while we load your content...

