
Switching charge kinetics from type-I to Z-scheme for g-C3N4 and ZnIn2S4 by defective engineering for efficient and durable hydrogen evolution†
 *a
and
Yiguo
Su
*a
*a
and
Yiguo
Su
*a
Abstract
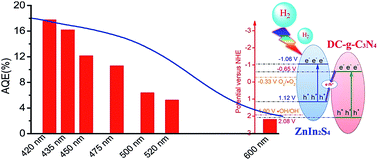
By virtue of the spatial separation of active sites, light harvesting as well as highly preserved redox capability, direct Z-scheme heterostructural photocatalysts are found as promising materials for solar energy conversion and environmental remediation. However, challenges still exist in regulating the electron flow direction between semiconductors with staggered electronic structures. In this regard, by regulating the defective crystalline features of g-C3N4, a direct Z-scheme DC-g-C3N4/ZnIn2S4 heterostructure was gained for the modulation of the electronic structure and robust hydrogen production performance. The insertion of defective groups into the carbon nitride matrix led to a drastic downshift of band edge potentials in comparison to that of pristine g-C3N4. This variation gave birth to a staggered band edge alignment between DC-g-C3N4 and ZnIn2S4, resulting in charge transfer kinetics variation from type-I to direct Z-scheme. By careful characterization, it was found that the highly crystalline DC-g-C3N4 coupled with ZnIn2S4 to show a fine interfacial contact. The optimal photocatalytic hydrogen evolution reaction (PHER) activity over DC-g-C3N4/ZnIn2S4 reached 1.65 mmol g−1 h−1 with an apparent quantum efficiency (AQE) of about 18.2% at 420 nm and an AQE of ∼2.2% at 600 nm. In combination with photocurrent measurements, photoluminescence spectra and electron paramagnetic resonance, the improved hydrogen evolution activity is regarded as the consequence of the decreased onset potential and improved spatial segregation of charge carriers via a direct Z-scheme carrier migration, where photoinduced electrons in DC-g-C3N4 can quickly combine with photoinduced holes in the valence band of ZnIn2S4, leading to the spatial separation of photoinduced electrons and holes between the two semiconductors.
 Please wait while we load your content...
Please wait while we load your content...

