
A pencil-and-paper method for elucidating halide double perovskite band structures†

Abstract
Halide double perovskites are an important emerging alternative to lead-halide perovskites in a variety of optoelectronic applications. Compared to ABX3 single perovskites (A = monovalent cation, X = halide), A2BB′X6 double perovskites exhibit a wider array of compositions and electronic structures, promising finer control over physical and electronic properties through synthetic design. However, a clear understanding of how chemical composition dictates the electronic structures of this large family of materials is still lacking. Herein, we develop a qualitative Linear Combination of Atomic Orbitals (LCAO) model that describes the full range of band structures for double perovskites. Our simple model allows for a direct connection between the inherently local bonding between atoms in the double perovskite and the resulting delocalized bands of the solid. In particular, we show how bands in halide double perovskites originate from the molecular orbitals of metal–hexahalide coordination complexes and describe how these molecular orbitals vary within a band. Our results provide both an enhanced understanding of known perovskite compositions and predictive power for identifying new compositions with targeted properties. We present a table, which permits the position of the conduction band minimum and valence band maximum in most double perovskites to be immediately determined from the frontier atomic orbitals of the B-site metals. Using purely qualitative arguments based on orbital symmetries and their relative energies, the direct/indirect nature of the bandgap of almost all halide double perovskites can thus be correctly predicted. We hope that this theory provides an intuitive understanding of halide double perovskite band structures and enables lessons from molecular chemistry to be applied to these extended solids.
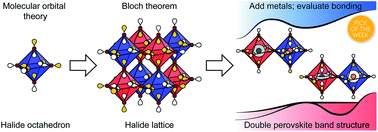
- This article is part of the themed collections: Perovskites march on, 2019 Chemical Science HOT Article Collection and 2019 ChemSci Pick of the Week Collection
 Please wait while we load your content...
Please wait while we load your content...

