
Mining predicted crystal structure landscapes with high throughput crystallisation: old molecules, new insights†

Abstract
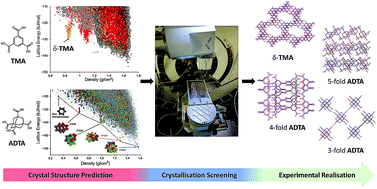
Organic molecules tend to close pack to form dense structures when they are crystallised from organic solvents. Porous molecular crystals defy this rule: they contain open space, which is typically stabilised by inclusion of solvent in the interconnected pores during crystallisation. The design and discovery of such structures is often challenging and time consuming, in part because it is difficult to predict solvent effects on crystal form stability. Here, we combine crystal structure prediction (CSP) with a robotic crystallisation screen to accelerate the discovery of stable hydrogen-bonded frameworks. We exemplify this strategy by finding new phases of two well-studied molecules in a computationally targeted way. Specifically, we find a new ‘hidden’ porous polymorph of trimesic acid, δ-TMA, that has a guest-free hexagonal pore structure, as well as three new solvent-stabilized diamondoid frameworks of adamantane-1,3,5,7-tetracarboxylic acid (ADTA). Beyond porous solids, this hybrid computational–experimental approach could be applied to a wide range of materials problems, such as organic electronics and drug formulation.
- This article is part of the themed collections: Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2020, Most popular 2019-2020 inorganic, main group and crystal engineering chemistry articles and Accelerating Chemistry Symposium Collection
 Please wait while we load your content...
Please wait while we load your content...

