
Configuration determination by residual dipolar couplings: accessing the full conformational space by molecular dynamics with tensorial constraints†

Abstract
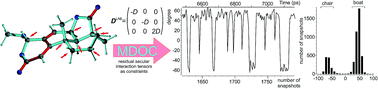
Residual dipolar couplings (RDCs) and other residual anisotropic NMR parameters provide valuable structural information of high quality and quantity, bringing detailed structural models of flexible molecules in solution in reach. The corresponding data interpretation so far is directly or indirectly based on the concept of a molecular alignment tensor, which, however, is ill-defined for flexible molecules. The concept is typically applied to a single or a small set of lowest energy structures, ignoring the effect of vibrational averaging. Here, we introduce an entirely different approach based on time averaged molecular dynamics with dipolar couplings as tensorial orientational restraints that can be used to solve structural problems in molecules of any size without the need of introducing an explicit molecular alignment tensor into the computation. RDC restraints are represented by their full 3D interaction tensor in the laboratory frame, for which pseudo forces are calculated using a secular dipolar Hamiltonian as the target. The resulting rotational averaging of each individual tensorial restraint leads to structural ensembles that best fulfil the experimental data. Using one-bond RDCs, the approach has been implemented in the force field procedures of the program COSMOS and extensively tested. A concise theoretical introduction, including the special treatment of force fields for stable and fast MD simulations, as well as applications regarding configurational analyses of small to medium-sized organic molecules with different degrees of flexibility, is given. The observed results are discussed in detail.
- This article is part of the themed collection: 2019 Chemical Science HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

