
Insights into the role of noncovalent interactions in distal functionalization of the aryl C(sp2)–H bond†
 a
and
Raghavan B.
Sunoj
*a
a
and
Raghavan B.
Sunoj
*a
Abstract
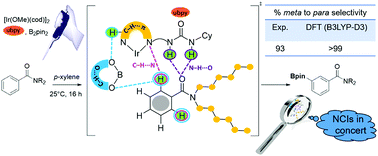
Burgeoning interest in distal functionalization of aryl C–H bonds led to the development of iridium-catalyzed borylation reactions. The significance and inadequate mechanistic understanding of C(sp2)–H borylations motivated us to investigate the key catalytic steps and the origin of a directing-group-free regiocontrol in the reaction between aryl amides and B2pin2 (bis(pinacolato)diboron). An Ir(III)(ubpy)tris(boryl) complex, generated from the pre-catalyst [Ir(OMe)(cod)]2 by the action of a bipyridine-urea ligand (ubpy) and B2pin2, is considered as the most likely active catalyst. The meta C–H activation of N,N-dihexylbenzamide is energetically more favorable over the para isomer. The origin of this preference is traced to the presence of a concerted action of noncovalent interactions (NCIs), primarily between the catalyst and the substrate, in the regiocontrolling transition states (TSs). Molecular insights into such TSs revealed that the N–H⋯O interaction between the tethered urea moiety of the Ir-bound ubpy ligand of the catalyst and the amide carbonyl of the substrate is a critical interaction that helps orient the meta C–H bond nearer to iridium. Other NCIs such as C–H⋯π between the substrate and the catalyst, C–H⋯O involving the substrate C–H and the oxygen of the B2pin2 ligand and C–H⋯N between the substrate and the N atom of the Ir-bound ubpy confirm the significance of such interactions in providing the desirable differential energies between the competing TSs that form the basis of the extent of regioselectivity.
- This article is part of the themed collections: Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2021, Celebrating the Chemical Sciences in India - Leaders in the Field Symposium 2020, Most popular 2019-2020 catalysis articles and Celebrating the Chemical Science in India - Leaders in the Field Symposium
 Please wait while we load your content...
Please wait while we load your content...

