
Protein–protein binding pathways and calculations of rate constants using fully-continuous, explicit-solvent simulations†

Abstract
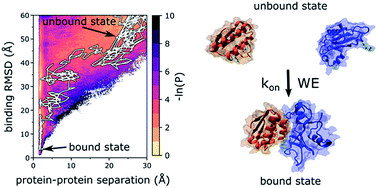
A grand challenge in the field of biophysics has been the complete characterization of protein–protein binding processes at atomic resolution. This characterization requires the direct simulation of binding pathways starting from the initial, unbound state and proceeding through states that are too transient to be captured by experiment. Here, we applied the weighted ensemble path sampling strategy to orchestrate atomistic simulation of protein–protein binding pathways. Our simulation generated 203 fully-continuous and independent pathways along with rate constants for the binding process involving the barnase and barstar proteins. Results reveal multiple binding pathways along a “funnel-like” free energy landscape in which the formation of the “encounter complex” intermediate is rate-limiting followed by a relatively rapid rearrangement of the encounter complex to the bound state. Among all diffusional collisions, only ∼11% were productive. In the most probable binding pathways, the proteins rotated to a large extent (likely via electrostatic steering) in order to collide productively followed by “rolling” of the proteins along each other's binding interfaces to reach the bound state. Consistent with experiment, R59 was identified as the most kinetically important barnase residue for the binding process. Furthermore, protein desolvation occurs late in the binding process during the rearrangement of the encounter complex to the bound state. Notably, the positions of crystallographic water molecules that bridge hydrogen bonds between barnase and barstar are occupied in the bound-state ensemble. Our simulation was completed in a month using 1600 CPU cores at a time, demonstrating that it is now practical to carry out atomistic simulations of protein–protein binding.
 Please wait while we load your content...
Please wait while we load your content...

