
Enabling synthesis in fragment-based drug discovery by reactivity mapping: photoredox-mediated cross-dehydrogenative heteroarylation of cyclic amines†
 *a
Tom D.
Heightman,
a
Steven V.
Ley,
b
Fabio
Lima
bc
and
Christopher N.
Johnson
*a
*a
Tom D.
Heightman,
a
Steven V.
Ley,
b
Fabio
Lima
bc
and
Christopher N.
Johnson
*a
Abstract
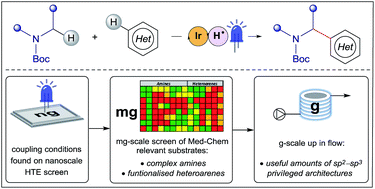
In fragment-based drug discovery (FBDD), a weakly binding fragment hit is elaborated into a potent ligand by bespoke functionalization along specific directions (growth vectors) from the fragment core in order to complement the 3D structure of the target protein. This structure-based design approach can present significant synthetic challenges, as growth vectors often originate on sp2 or sp3 ring carbons which are not the most synthetically accessible points on the fragment. To address this issue and expedite synthesis in FBDD, we established a nanogram-to-gram workflow for the development of enabling synthetic transformations, such as the direct C–H functionalization of heterocycles. This novel approach deploys high-throughput experimentation (HTE) in 1536-well microtiter plates (MTPs) facilitated by liquid handling robots to screen reaction conditions on the nanomolar scale; subsequently the reaction is upscaled in a continuous flow to generate gram-quantities of the material. In this paper, we disclose the use of this powerful workflow for the development of a photoredox-mediated cross-dehydrogenative coupling of fragments and medicinally relevant heterocyclic precursors via Minisci-type addition of α-amino radicals to electron-deficient heteroarenes. The optimized reaction conditions were employed on the milligram-scale on a diverse set of 112 substrates to map out structure–reactivity relationships (SRRs) of the transformation. The coupling exhibits excellent tolerance to a variety of functional groups and N-rich heteroarenes relevant to FBDD and was upscaled in a continuous flow to afford gram-quantities of pharmaceutically relevant sp2–sp3 privileged architectures.
- This article is part of the themed collection: Most popular 2019-2020 organic chemistry articles
 Please wait while we load your content...
Please wait while we load your content...

