
Supramolecular cage encapsulation as a versatile tool for the experimental quantification of aromatic stacking interactions†

Abstract
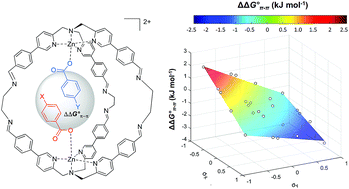
The widespread presence of aromatic stacking interactions in chemical and biological systems, combined with their relatively small energetic contribution, have led to a plethora of theoretical and experimental studies for their quantification and rationalization. Typically, π–π aromatic interactions are studied as a function of substituents to gather information about the interaction mechanism. While experiments suggest that aromatic interactions are dominated by local electrostatic contacts between π-electron density and CH groups, theoretical work has raised the possibility that direct electrostatic interactions between local dipoles of the substituents may play a role. We describe a supramolecular cage that binds two aromatic carboxylates in a stacked geometry such that the aromatic substituents are remote in space. Chemical Double Mutant Cycles (DMCs) were used to measure fifteen different aromatic stacking interactions as a function of substituent (NMe2, OMe, Me, Cl and NO2). When both aromatic rings have electron-withdrawing nitro substituents, the interaction is attractive (−2.8 kJ mol−1) due to reduced π-electron repulsion. When both aromatic rings have electron-donating di-methylamino substituents, the interaction is repulsive (+2.0 kJ mol−1) due to increased π-electron repulsion. The results show that aromatic stacking interactions are dominated by short range electrostatic contacts rather than substituent dipole interactions.
- This article is part of the themed collection: 1st International Conference on Noncovalent Interactions
 Please wait while we load your content...
Please wait while we load your content...

