
A graph-convolutional neural network model for the prediction of chemical reactivity†

Abstract
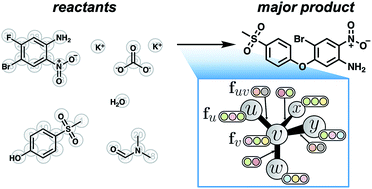
We present a supervised learning approach to predict the products of organic reactions given their reactants, reagents, and solvent(s). The prediction task is factored into two stages comparable to manual expert approaches: considering possible sites of reactivity and evaluating their relative likelihoods. By training on hundreds of thousands of reaction precedents covering a broad range of reaction types from the patent literature, the neural model makes informed predictions of chemical reactivity. The model predicts the major product correctly over 85% of the time requiring around 100 ms per example, a significantly higher accuracy than achieved by previous machine learning approaches, and performs on par with expert chemists with years of formal training. We gain additional insight into predictions via the design of the neural model, revealing an understanding of chemistry qualitatively consistent with manual approaches.
- This article is part of the themed collections: Most popular 2019-2020 physical and theoretical chemistry articles, Accelerating Chemistry Symposium Collection, Most popular 2018-2019 physical and theoretical chemistry articles and The ChemRxiv Collection
 Please wait while we load your content...
Please wait while we load your content...

