
Hot-electron effects during reactive scattering of H2 from Ag(111): the interplay between mode-specific electronic friction and the potential energy landscape†

Abstract
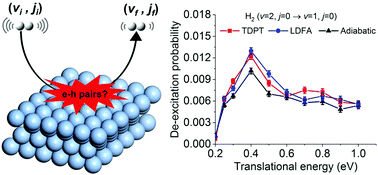
The breakdown of the Born–Oppenheimer approximation gives rise to nonadiabatic effects in gas-surface reactions at metal surfaces. However, for a given reaction, it remains unclear which factors quantitatively determine whether these effects measurably contribute to surface reactivity in catalysis and photo/electrochemistry. Here, we systematically investigate hot electron effects during H2 scattering from Ag(111) using electronic friction theory. We combine first-principles calculations of tensorial friction by time-dependent perturbation theory based on density functional theory and an analytical neural network representation, to overcome the limitations of existing approximations and explicitly simulate mode-specific nonadiabatic energy loss during molecular dynamics. Despite sizable hot-electron-induced energy loss, no measurable nonadiabatic effects can be found for H2 scattering on Ag(111). This is in stark contrast to previous reports for vibrationally excited H2 scattering on Cu(111). By detailed analysis of the two systems, we attribute this discrepancy to a subtle interplay between the magnitude of electronic friction along intramolecular vibration and the shape of the potential energy landscape that controls the molecular velocity at impact. On the basis of this characterization, we offer guidance for the search of highly nonadiabatic surface reactions.
 Please wait while we load your content...
Please wait while we load your content...

