
Decomposition kinetics for HONO and HNO2
 †b
and
C.
Franklin Goldsmith
*b
†b
and
C.
Franklin Goldsmith
*b
Abstract
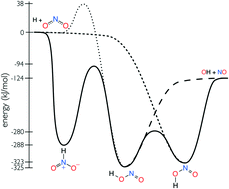
This work presents a detailed investigation into the isomerization and decomposition of HONO and HNO2. State-of-the-art electronic structure theory is used to compute the HNO2 potential energy surface. Temperature and pressure dependent rate coefficients are computed using microcanonical rate theory and the master equation. The electronic structure theory properties are optimized against the relevant experimental data. A novel strategy was developed to incorporate uncertainty in the minimum energy pathway into the optimized mechanism. The new mechanism is in excellent agreement with all available experimental data for H + NO2 → OH + NO and OH + NO → HONO. The calculations identify OH + NO as the dominant products for HNO2, which were neglected from all previous mechanisms in the literature.
- This article is part of the themed collection: Reaction Chemistry & Engineering Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

