
Middle-down approach: a choice to sequence and characterize proteins/proteomes by mass spectrometry†

Abstract
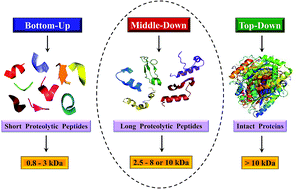
Owing to rapid growth in the elucidation of genome sequences of various organisms, deducing proteome sequences has become imperative, in order to have an improved understanding of biological processes. Since the traditional Edman method was unsuitable for high-throughput sequencing and also for N-terminus modified proteins, mass spectrometry (MS) based methods, mainly based on soft ionization modes: electrospray ionization and matrix-assisted laser desorption/ionization, began to gain significance. MS based methods were adaptable for high-throughput studies and applicable for sequencing N-terminus blocked proteins/peptides too. Consequently, over the last decade a new discipline called ‘proteomics’ has emerged, which encompasses the attributes necessary for high-throughput identification of proteins. ‘Proteomics’ may also be regarded as an offshoot of the classic field, ‘biochemistry’. Many protein sequencing and proteomic investigations were successfully accomplished through MS dependent sequence elucidation of ‘short proteolytic peptides (typically: 7–20 amino acid residues), which is called the ‘shotgun’ or ‘bottom-up (BU)’ approach. While the BU approach continues as a workhorse for proteomics/protein sequencing, attempts to sequence intact proteins without proteolysis, called the ‘top-down (TD)’ approach started, due to ambiguities in the BU approach, e.g., protein inference problem, identification of proteoforms and the discovery of posttranslational modifications (PTMs). The high-throughput TD approach (TD proteomics) is yet in its infancy. Nevertheless, TD characterization of purified intact proteins has been useful for detecting PTMs. With the hope to overcome the pitfalls of BU and TD strategies, another concept called the ‘middle-down (MD)’ approach was put forward. Similar to BU, the MD approach also involves proteolysis, but in a restricted manner, to produce ‘longer’ proteolytic peptides than the ones usually obtained in BU studies, thereby providing better sequence coverage. In this regard, special proteases (OmpT, Sap9, IdeS) have been used, which can cleave proteins to produce longer proteolytic peptides. By reviewing ample evidences currently existing in the literature that is predominantly on PTM characterization of histones and antibodies, herein we highlight salient features of the MD approach. Consequently, we are inclined to claim that the MD concept might have widespread applications in future for various research areas, such as clinical, biopharmaceuticals (including PTM analysis) and even for general/routine characterization of proteins including therapeutic proteins, but not just limited to analysis of histones or antibodies.
 Please wait while we load your content...
Please wait while we load your content...

