
Theoretical insight into the photophysical properties of six heteroleptic Ir(iii) phosphorescent complexes bearing ppy-type ligands†
 ab
ab
Abstract
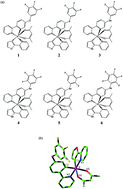
By using density functional theory and time-dependent density functional theory, the geometrical, electronic and photophysical properties of six complexes with two ppy-type ligands and one acetylacetone anion around the Ir center have been explored. The lowest energy absorption wavelengths are located at 414 nm for 1, 434 nm for 2, 434 nm for 3, 421 nm for 4, 436 nm for 5, and 425 nm for 6, respectively. The lowest energy emissions of these complexes are localized at 617, 492, 633, 634, 491 and 491 nm, respectively, for complexes 1–6, simulated in CH2Cl2 medium at the M062X level. The calculated lowest lying absorption wavelength and the lowest energy emission wavelength for complex 3 are very close to the available experimental values. The position and number of the incorporated electron-withdrawing fluorine substituents have some effect on the electronic and photophysical properties of these studied complexes.
 Please wait while we load your content...
Please wait while we load your content...