
Computer modeling of 2D supramolecular nanoporous monolayers self-assembled on graphite†
 *ade
Fabrice
Charra
f
and
*ade
Fabrice
Charra
f
and
Abstract
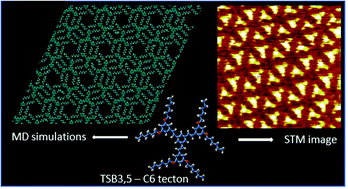
Nano-porous two-dimensional molecular crystals, self-assembled on atomically flat host surfaces offer a broad range of possible applications, from molecular electronics to future nano-machines. Computer-assisted designing of such complex structures requires numerically intensive modeling methods. Here we present the results of extensive, fully atomistic simulations of self-assembled monolayers of interdigitated molecules of 1,3,5-tristyrilbenzene substituted by C6 alkoxy peripheral chains (TSB3,5-C6), deposited onto highly-ordered pyrolytic graphite. Structural and electronic properties of the TSB3,5-C6 molecules were determined from ab initio calculations, then used in Molecular Dynamics simulations to analyze the mechanism of formation, epitaxy, and stability of the TSB3,5-C6 nanoporous superlattice. We show that the monolayer disordering results from the competition between flexibility of the C6 chains and their stabilization by interdigitation. The inclusion of guest molecules (benzene and pyrene) into superlattice nanopores stabilizes the monolayer. The alkoxy chain mobility and available pore space defines the systems dynamics, essential for potential application.
 Please wait while we load your content...
Please wait while we load your content...

