
An experimental and theoretical study of metallorganic coordination networks of tetrahydroxyquinone on Cu(111)†
 *a
Matteo
Lo Cicero,
bc
Carlo
Mariani,
e
Maria Grazia
Betti,
e
Mauro
Sambi
af
and
Maurizio
Casarin
*a
*a
Matteo
Lo Cicero,
bc
Carlo
Mariani,
e
Maria Grazia
Betti,
e
Mauro
Sambi
af
and
Maurizio
Casarin
*a
Abstract
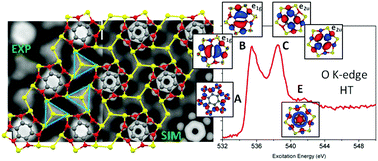
The adsorption of tetrahydroxyquinone (THQ) on the Cu(111) surface at different temperatures gives rise to different ordered structures, unveiled by density functional theory (DFT) modelling of scanning tunnelling microscopy (STM) topography and by X-ray absorption spectroscopy (XAS) measurements. After annealing, the THQ films generated tetrameric Cu clusters embedded in a metal–organic tetraoxyquinone (TOQ) network, whose electronic properties were thoroughly studied. The remarkable agreement between the STM results and periodic calculations was exploited to model C and O K-edge XA spectra recorded before (room temperature, RT) and after annealing (high temperature, HT). The subsequent analysis of the C and O TOQ@Cu(111) K-edge XA spectra provides definitive information about the structural arrangement of the TOQ@Cu(111) coordination network, confirming the complete THQ dehydrogenation upon annealing, suggested by the simulations of the STM images. The obtained information is crucial to rationalize the evolution of THQ@Cu(111) upon heating, shedding light into the potential reactivity of the HT coordination network, and, more generally, to investigate the nature and character of low-lying virtual states, the most involved states in the chemical reactions.
 Please wait while we load your content...
Please wait while we load your content...

