
Oxygen reduction reaction mechanism on P, N co-doped graphene: a density functional theory study†

Abstract
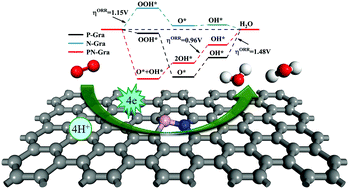
Recently, heteroatom co-doped carbon material catalysts have shown extraordinary oxygen reduction reaction (ORR) catalytic activity, but a lack of understanding of the specific mechanism of the ORR has hindered their further development. In this study, we studied the geometric structure, stability, electronic properties, catalytic sites and detailed ORR pathways of P, N co-doped graphene (PN-Gra) by density functional theory (DFT) calculations, trying to reveal the effect of N and P co-doped graphene on the catalytic activity of the ORR. Our calculations indicate that the PN-Gra structure remains stable at high temperature (1000 K) and P, N atoms with their adjacent four carbon atoms constitute the active region to adsorb O2 more efficiently than single phosphorus or nitrogen doped catalysts. Combining energy barrier and free energy diagram analysis, the most favorable pathway is O2* → O* + O* → OH* + O* → 2OH* → OH* + H2O → 2H2O, which is thermodynamically favorable and the associated energy barrier is low, with an overpotential of 0.96 V. After comprehensive analysis and comparison, P, N co-doping does improve the catalytic ability due to the synergistic effect compared to single doping. We believe that this study provides researchers a way to understand the reaction mechanism and ways to improve the catalytic properties of carbon-based materials.
 Please wait while we load your content...
Please wait while we load your content...

