
Computational insight into a new family of functionalized tetrazole-N-oxides as high-energy density materials†
 *a
Jianguo
Zhang
c
*a
Jianguo
Zhang
c
Abstract
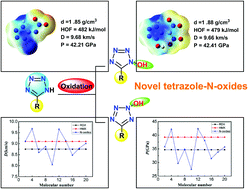
A new family of tetrazole-N-oxides comprising the energetic N–O moiety and the tetrazole skeleton with functionalities was designed based on density functional theory (DFT) calculations. The optimized geometry, electronic properties, IR spectra and thermodynamics were calculated first. Their energetic parameters including density, heats of formation, detonation properties and impact sensitivity were extensively evaluated. These newly designed tetrazole-N-oxides exhibit moderate impact sensitivities, high density (up to 1.89 g cm−3), good heats of formation (468.59–944.64 kJ mol−1), and excellent detonation performance, which in some cases (D = 9.68 km s−1, P = 42.21 GPa; D = 9.66 km s−1, P = 42.41 GPa) outperform the current explosive benchmark HMX, making them promising candidates for new environmentally friendly high-energy density compounds. This functionalized tetrazole ring with the N–O moiety system results in a compromise between the desirable stabilities and high detonation properties, and thus may enhance future utilization in the design and synthesis of new energetic materials.
 Please wait while we load your content...
Please wait while we load your content...

