
Theoretical study of the substrate and molecular density effects on molecular self-assembly†
 a
a
Abstract
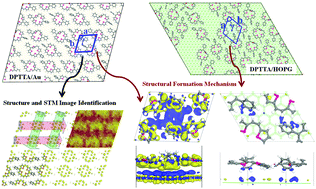
This work is aiming at theoretically exploring the substrate and molecular density effects on molecular self-assembly. Density functional theory (DFT) and molecular dynamics (MD) simulations were used to investigate the atomic arrangement of a sulfur-bridged annulene (i.e., DPTTA) on both Au and highly oriented pyrolytic graphite (HOPG) substrates. The critical molecular density of the DPTTA/Au self-assembled structure was determined to be 0.52 nm−2 by the construction and simulation of different DPTTA molecular density models. By extracting and optimizing the self-assembled unit, clear identification of the self-assembled atomic structure and the STM image were realized. It was found that DPTTA molecules cannot form 2D self-assembled structures on a HOPG substrate. The reason for the different configurations of DPTTA/Au and DPTTA/HOPG systems was successfully revealed by the calculations of adsorption energy, weak interaction energy and electron density differences. This study provides a reference for the quantitative prediction of the self-assembled critical molecular density and the accurate determination of self-assembled atomic structures by theoretical simulation.
 Please wait while we load your content...
Please wait while we load your content...

