
Mechanistic insights and computational design of half-sandwich iridium and rhodium complexes for hydrogenation of quinoline†
 ab
and
Xinzheng
Yang
*ab
ab
and
Xinzheng
Yang
*ab
Abstract
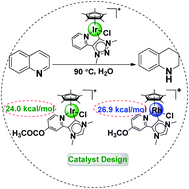
A detailed reaction mechanism of the hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline catalysed by a half-sandwich Cp*Ir triazolylidene complex was computationally investigated by using density functional theory. The direct proton transfer from Ir to the nitrogen atom in quinoline was found to be the rate-determining step with a total barrier of 25.1 kcal mol−1 in free energy. Furthermore, by replacing the hydrogen atoms at the para (R) positions of the pyridine ligand with different functional groups, and the triazolylidene ligands with various N-heterocyclic carbenes, we proposed a series of half-sandwich Ir, Rh, and Co complexes and computationally screened four promising candidates for the catalytic hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline with predicted total free energy barriers close to 24.0 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

