
Single-atom Pt on non-metal modified graphene sheets as efficient catalysts for CO oxidation†
 ‡*a
‡*a
Abstract
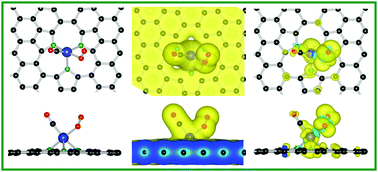
By the density functional theory (DFT) calculations, the formation geometries, electronic structures and catalytic properties of metal Pt and nonmetal (NM) atom-co-modified graphene (Pt–3NM–graphene, NM = N, Si, P) as reactive substrates were investigated. First, the formation energy of the Pt–3N–graphene configuration was less than that of the Pt–3Si– and Pt–3P–graphene systems. The adsorbed O2 on Pt–3NM–graphene was more stable than that of CO molecules and their corresponding electronic and magnetic properties were analyzed in detail. Compared with the isolated O2 or CO molecule, the coadsorption of CO/O2 (or 2CO) had larger adsorption energies on Pt–3NM–graphene, which might have facilitated the catalytic reactions for CO oxidation. Furthermore, the different reaction mechanisms of CO oxidation on Pt–3NM–graphene were systematically investigated. It was found that the Eley–Rideal (ER) mechanism (CO + O2 → CO3), as the initial state on Pt–3NM–graphene sheets, had larger energy barriers than those of the Langmuir–Hinshelwood (LH) and new termolecular Eley–Rideal (TER) mechanisms. For the Pt–3N– and Pt–3Si–graphene, the catalytic oxidation of CO reactions through LH and TER reactions had much small energy barriers (<0.3 eV), which indicated that the initial state was energetically more favorable. These results provide valuable guidance on selecting dopants in graphene to design carbon-based catalysts with low price and high activity.
 Please wait while we load your content...
Please wait while we load your content...

