
A network-based approach reveals novel invasion and Maurer's clefts-related proteins in Plasmodium falciparum†
 *a
and
Gopalakrishnan
Bulusu
*a
*a
and
Gopalakrishnan
Bulusu
*a
Abstract
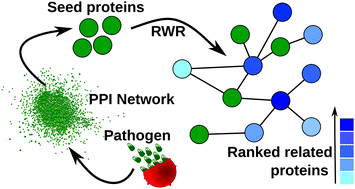
Malaria continues to be a major concern in developing countries despite continuous efforts to find a cure for the disease. Understanding the pathogenesis mechanism is necessary to identify more effective drug targets against malaria. Many years of experimental research have generated a large amount of data for the malarial parasite, Plasmodium falciparum. These data are useful to understand the importance of certain parasite proteins, but it often remains unclear how these proteins come together, interact with other proteins and carry out their function. Identification of all proteins involved in pathogenesis is an important step towards understanding the molecular mechanism of pathogenesis. In this study, dynamic stage-specific protein–protein interaction networks were created based on gene expression data during the parasite's intra-erythrocytic stages and static protein–protein interaction data. Using previously known proteins of a biological event as seed proteins, the random walk with restart (RWR) method was used on the dynamic protein–protein interaction networks to identify novel proteins related to that event. Two screening procedures namely, permutation test and GO enrichment test were performed to increase the reliability of the RWR predictions. The proposed method was first validated on Plasmodium falciparum proteins related to invasion, where it could reproduce the existing knowledge from a small set of seed proteins. It was then used to identify novel Maurer's clefts resident proteins, where it could identify 152 parasite proteins. We show that the current approach can annotate conserved proteins with unknown function. The predicted proteins can help build a mechanistic model for disease pathogenesis, which will be useful in identifying new drug targets.
 Please wait while we load your content...
Please wait while we load your content...