
Antibody–antigen complex modelling in the era of immunoglobulin repertoire sequencing†

Abstract
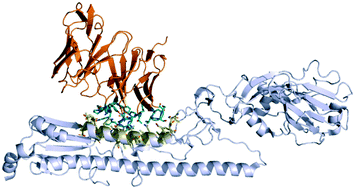
The natural immune repertoire can be a useful guide to antibody discovery against any given target. However, the large volume of immunoglobulin gene sequencing data necessitates the rational prioritisation of possible binders for experimental validation. Where other known binders exist, sequence similarity is used to infer binding, but this neglects alternative binding modes to the same epitope, and cannot identify antibodies that bind to different epitopes. In this review, we summarise the state-of-the-art of high-throughput antibody–antigen complex modelling. Given the millions of natural antibody sequences now available, this pipeline attempts to predict whether, and if so how, each antibody binds to a particular antigen's surface. We cover the current paradigm (antibody and antigen structural modelling, followed by binding site prediction, followed by molecular docking), discussing how existing algorithms can deal with this magnitude of data by balancing accuracy with computational efficiency, and identifying areas where further developments are required to improve performance.
- This article is part of the themed collection: Engineering immunity with quantitative tools
 Please wait while we load your content...
Please wait while we load your content...

