
Efforts in redesigning the antileukemic drug 6-thiopurine: decreasing toxic side effects while maintaining efficacy†

Abstract
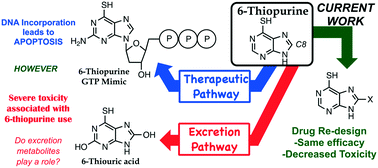
6-Thiopurine (6TP) is a currently prescribed drug in the treatment of diseases ranging from Crohn's disease to acute lymphocytic leukemia. While its potent mode of action is through incorporation into DNA as a thiol mimic of deoxyguanosine, severe toxicities are associated with its administration which hinder the potential therapeutic application. We have previously reported in vitro that the oxidative metabolites of 6TP, specifically 6-thiouric acid (6TU, Ki 7 μM), are potent inhibitors of UDP-glucose dehydrogenase (UDPGDH), an enzyme that is responsible for the formation of UDP-glucuronic acid (UDPGA), an essential substrate that is used in detoxification processes in the liver. An in vivo investigation was undertaken to probe if 6TU inhibits UDPGDH in rat hepatocytes, and it was observed that 6TU does greatly suppress the conjugation of bilirubin with UDPGA. The failed excretion of bilirubin is linked to a majority of the reported toxicities associated with 6TP administration. Efforts were undertaken for the construction of 6TP analogs, substituted at the C8 position, to reduce inhibition of UDPGDH while retaining therapeutic efficacy. Three new 6TP analogs bearing a halogen (Br, Cl, and F) at the C8 position have been achieved over five-synthetic steps in overall yields of 16 to 32%. Each of these analogs were shown to have reduced inhibition towards UDPGDH, with Ki values of 192, 163, 215 μM, respectively. In addition, the bromine, chlorine, and fluorine analogs were shown to possess cytotoxicity towards the REH cell line (acute lymphocytic leukemia) having IC50 values of 9.54 μM (±0.97), 3.95 μM (±1.94), and 4.71 μM (±1.40), respectively. These three new 6TP analogs represent the first steps in the redesign of this potent anticancer agent into a better drug that possesses reduced toxic side effects while retaining therapeutic potency.
 Please wait while we load your content...
Please wait while we load your content...