
Temperature dependence of the vibrational spectrum of porphycene: a qualitative failure of classical-nuclei molecular dynamics†
 b
and
Mariana
Rossi
*a
b
and
Mariana
Rossi
*a
Abstract
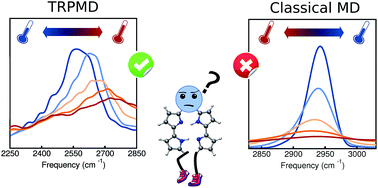
The temperature dependence of vibrational spectra can provide information about structural changes of a system and also serve as a probe to identify different vibrational mode couplings. Fully anharmonic temperature-dependent calculations of these quantities are challenging due to the cost associated with statistically converging trajectory-based methods, especially when accounting for nuclear quantum effects. Here, we train a high-dimensional neural network potential energy surface for the porphycene molecule based on data generated with DFT-B3LYP, including pairwise van der Waals interactions. In addition, we fit a kernel ridge regression model for the molecular dipole moment surface. The combination of this machinery with thermostatted path integral molecular dynamics (TRPMD) allows us to obtain well-converged, full-dimensional, fully-anharmonic vibrational spectra including nuclear quantum effects, without sacrificing the first-principles quality of the potential-energy surface or the dipole surface. Within this framework, we investigate the temperature and isotopologue dependence of the high-frequency vibrational fingerprints of porphycene. While classical-nuclei dynamics predicts a red shift of the vibrations encompassing the NH and CH stretches, TRPMD predicts a strong blue shift in the NH-stretch region and a smaller one in the CH-stretch region. We explain this behavior by analyzing the modulation of the effective potential with temperature, which arises from vibrational coupling between quasi-classical thermally activated modes and high-frequency quantized modes.
- This article is part of the themed collection: Quantum effects in complex systems
 Please wait while we load your content...
Please wait while we load your content...

