
Direct dynamics analysis of the cationic Cp*(PMe3)Ir(CH3) methane C–H activation mechanism

Abstract
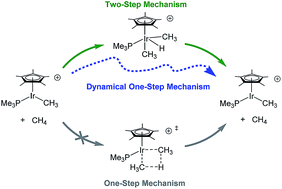
For the σ-bond metathesis reaction between methane and cationic Cp*(PMe3)IrIII(CH3), previous static DFT calculations revealed a two-step oxidative addition/reductive elimination mechanism with an intervening IrV–H intermediate. We recently reported quasiclassical direct molecular dynamics simulations where starting from the vibrationally-averaged oxidative addition transition state a minor, but significant, amount of trajectories bypassed the IrV–H intermediate in a dynamical one-step mechanism. These trajectories also revealed that after the reductive coupling transition state is passed methane always dissociates and the C–H σ-complex is skipped. Here we report direct dynamics simulations using a microcanonical temperature sampling method with trajectories initialized and propagated using the Gaussian program. Sets of 30 trajectories were examined with average time steps ranging from 1.5 to 0.25 femtoseconds (fs). These results showed that a step size of ∼1.5 fs is too large and likely overestimates the amount of trajectories skipping the IrV–H intermediate. Trajectories with 0.75 and 0.25 fs time steps gave qualitatively similar profiles compared to our previous results using our program DynSuite with a 1 fs time step. Reverse trajectories with an average time step of 0.25 fs showed complete skipping of the methane σ complex. 30 trajectories were also propagated using a SMD continuum dichloromethane solvent model. These trajectories showed very similar behavior to gas-phase trajectories.
- This article is part of the themed collection: Mechanistic processes in organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

