
Neutral, cationic and anionic organonickel and -palladium complexes supported by iminophosphine/phosphinoenaminato ligands†‡

Abstract
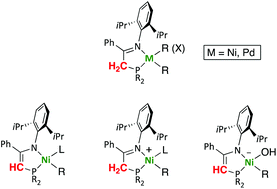
We report a series of organometallic nickel and palladium complexes containing iminophosphine ligands R2PCH2C(Ph) = N-Dipp (Dipp = 2,6-diisopropylphenyl; R = iPr, La; R = Ph, Lb; and R = o-C6H4OMe, Lc), synthesized by ligand exchange or oxidative addition reactions, and we investigate the capacity of such ligands to undergo reversible deprotonation to the corresponding phosphinoenaminato species. In the attempted ligand exchange reaction of the nickel bis(trimethylsilyl)methyl precursor [Ni(CH2SiMe3)2Py2] with Lb, the iminophosphine acts as a weak acid rather than a neutral ligand, cleaving one of the Ni–C bonds, to afford the phosphinoenaminato complex [Ni(CH2SiMe3)(L′b)(Py)] (L′b = conjugate base of Lb). We disclose a general method for the syntheses of complexes [Ni(CH2SiMe3)(L)(Py)]+ (L = La, Lb or Lc), and demonstrate that iminophosphine deprotonation is a general feature and occurs reversibly in the coordination sphere of the metal. By studying proton exchange reactions of the cation [Ni(CH2SiMe3)(Lb)(Py)]+ with bases of different strength we show that the conjugate phosphinoenaminato ligand in [Ni(CH2SiMe3)(L′b)(Py)] is a base with strength comparable to DBU in THF. The acyl group in the functionalized aryl complex [Ni(p-C6H4COCH3)(Br)(La)] does not interfere in the iminophosphine deprotonation with NaH. The latter reaction affords the unusual anionic hydroxide species [Ni(p-C6H4COCH3)(OH)(L′a)]−Na+, which was isolated and fully characterized.
 Please wait while we load your content...
Please wait while we load your content...

