
Supermetal: SbF5-mediated methane oxidation occurs by C–H activation and isobutane oxidation occurs by hydride transfer†

Abstract
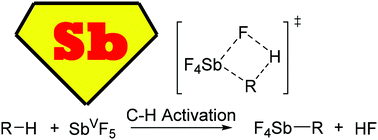
SbVF5 is generally assumed to oxidize methane through a methanium-to-methyl cation mechanism. However, experimentally no H2 is observed, and the mechanism of methane oxidation has remained unsolved for several decades. To solve this problem, density functional theory calculations with multiple chemical models (mononuclear and dinuclear) were used to examine methane oxidation by SbVF5 in the presence of CO leading to the methyl acylium cation ([CH3CO]+). While there is a low barrier for methane protonation by [SbVF6]−[H]+ (the combination of SbVF5 and HF) to give the [SbVF5]−[CH5]+ ion pair, H2 dissociation is a relatively high energy process, even with CO assistance, and so this protonation pathway is reversible. While Sb-mediated hydride transfer has a reasonable barrier, the C–H activation/σ-bond metathesis mechanism with the formation of an SbV–Me intermediate is lower in energy. This pathway leads to the acylium cation by functionalization of the SbV–Me intermediate with CO and is consistent with no observation of H2. Because this C–H activation/metal-alkyl functionalization pathway is higher in energy than methane protonation, it is also consistent with the experimentally observed methane hydrogen-to-deuterium exchange. This is the first time that evidence is presented demonstrating that SbVF5 acts beyond a Bronsted superacid and involves C–H activation with an organometallic intermediate. In contrast to methane, due to the much lower carbocation hydride affinity, isobutane significantly favors hydride transfer to give the tert-butyl carbocation with concomitant SbV to SbIII reduction. In this mechanism, the resulting highly acidic SbV–H intermediate provides a route to H2 through protonation of isobutane, which is consistent with experiments and resolves the longstanding enigma of different experimental results for methane versus isobutane.
 Please wait while we load your content...
Please wait while we load your content...

