
Experimental and theoretical investigation of lithium-ion conductivity in Li2LaNbTiO7†

Abstract
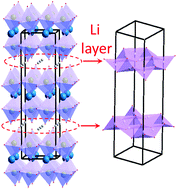
In line with efforts to discover new classes of lithium-conducting solids, this work utilizes density functional theory (DFT), neutron diffraction, and electrochemical impedance spectroscopy to study the lithium ion mobility in the new Ruddlesden–Popper-type oxide, Li2LaNbTiO7, where bilayer stacks of (Nb/Ti)O6 octahedra are separated by layers of lithium ions. In this material, the lithium hopping distances have been shortened by incorporation of ions with smaller ionic radii, leading to a pronounced improvement in Li-ion conductivity in this family of oxides. Further enhancement of Li-ion conductivity was achieved by creation of defects in the lithium layer through the synthesis of Li1.8LaNb1.2Ti0.8O7, which contains 10% lithium-deficiency. The defects facilitate the mobility of lithium ions, leading to greater Li-ion conductivity. Detailed analysis of real and imaginary components of impedance spectroscopy and dielectric properties highlight the impact of these strategies. Neutron diffraction helps with accurate determination of lithium positions given that lithium is nearly invisible to laboratory X-ray diffraction. DFT calculations show a wide band gap, and elucidate the direction of lithium diffusion in the material lattice, as well as the energy barriers associated with Li diffusion. These calculations show that lithium diffusion occurs parallel to b and c axes of the unit cell. They also show an energy bottleneck, which is a result of close proximity of lithium and oxygen in inter-stack spaces.
 Please wait while we load your content...
Please wait while we load your content...

