
Protonation and electrochemical properties of a bisphosphide diiron hexacarbonyl complex bearing amino groups on the phosphide bridge†
 b
and
Tsutomu
Mizuta
*a
b
and
Tsutomu
Mizuta
*a
Abstract
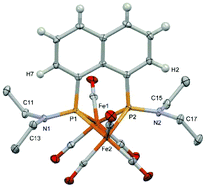
A bisphosphide-bridged diiron hexacarbonyl complex 3 with NEt2 groups on the phosphide bridge was synthesized to examine a new proton relay system from the NEt2 group to the bridging hydride between the two iron centers. As a precursor of the bridging moiety, peri-Et2NP–PNEt2-bridged naphthylene 5 was synthesized by the reaction of 1,8-dilithionaphthylene with two equivalents of Cl2PNEt2 followed by reductive P–P bond formation by magnesium. The reaction of the diphosphine ligand 5 with Fe2(CO)9 gave the diiron hexacarbonyl complex 3, in which the P–P bond of the ligand was cleaved to form the bisphosphide-bridge. The molecular structure of 3 indicated that the trigonal plane of the NEt2 group was forced to face the Fe–Fe bond to avoid steric congestion with the naphthylene group linking the two phosphide groups. The NEt2 group could be protonated by p-toluenesulfonic acid. Density functional theory (DFT) calculations confirmed that the proton of the N(H)Et2 group adopted a position close to the bridging hydride. The DFT results for the ferrocene analogue 1, in which the 1,8-naphthylene group of 3 was replaced with the 1,1′-ferrocenylene group, also revealed that the most stable orientation of the protonated NHEt2 group was that in the protonated 3. As a result, electrochemical proton reduction reactions using complexes 1 and 3 proceeded with similar catalytic efficiencies. Unfortunately, the catalytic efficiencies (CEs) of these complexes were much lower than those of the complexes with a proton relay system of the terminal hydrogen, indicating that the reactive properties of the bridging hydride in the present proton relay system cannot exceed those of the terminal hydride.
 Please wait while we load your content...
Please wait while we load your content...

