
Synthesis, structures, redox properties, and theoretical calculations of thiohalide capped octahedral hexanuclear technetium(iii) clusters†
 *ab
*ab
Abstract
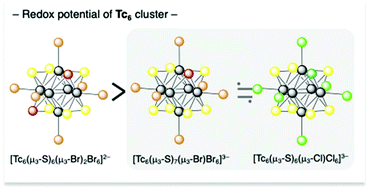
The first thiohalide μ3-capped octahedral hexanuclear technetium clusters with 24 valence electrons, [Tc6(μ3-S)8−n(μ3-Br)nBr6]n−4 [n = 1 ([Tc-S7Br]3−) and n = 2 ([Tc-S6Br2]2−)] and [Tc6(μ3-S)7(μ3-Cl)Cl6]3− ([Tc-S7Cl]3−), were synthesized and characterized. The structures of [Tc-S7Br]3−, [Tc-S6Br2]2−, and [Tc-S7Cl]3− were determined by single-crystal X-ray analysis. The Tc–Tc bond distances in [Tc-S7Br]3−, [Tc-S6Br2]2−, and [Tc-S7Cl]3− are 2.5842(6)–2.6029(6) Å (avg. 2.593(2) Å), 2.5835(10)–2.6049(10) Å (avg. 2.596(1) Å), and 2.5829(4)–2.5940(4) Å (avg. 2.587(3) Å), respectively. The capping halide and sulfide ligands in [Tc-S7Br]3−, [Tc-S6Br2]2−, and [Tc-S7Cl]3− were disordered in the crystals. The bond distances of Tc–S/Br as a function of the occupancies of the capping bromides for [Tc-S6Br2]2−, [Tc-S7Br]3−, and [Tc6(μ3-S)8Br6]4− ([Tc-S8]4−) showed a linear correlation. The one-electron reduction waves assignable to the TcIII6/TcIITcIII5 [Tc6(24e/25e)] process were observed for the novel complexes. Density functional theory (DFT) calculations of the hexanuclear technetium complexes showed a smaller energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the hexanuclear technetium complexes compared to those of the rhenium analogues. The electronic transitions of the new technetium complexes shifted to lower energy compared to the isotypic rhenium complexes.
 Please wait while we load your content...
Please wait while we load your content...

